4 DNA extraction and purification
Learning Objectives
- To recognize the role of the different reagents (e.g., detergents, salts, enzymes) and understand their function in DNA isolation and purification.
- To learn to use standard laboratory equipment for DNA extraction, such as pipettes, centrifuges, microtubes, and bead-beaters.
- To practice accurate pipetting, mixing, and handling of biological samples.
- To visualize and confirm DNA extraction.
- To understand factors affecting DNA yield and purity.
- To discuss how extracted DNA can be used in downstream applications, such as PCR, sequencing, cloning, and genetic analysis, to reinforce the significance of a successful extraction process.
What is the purpose of extracting DNA from cells?
Extracting DNA from cells has multiple purposes in research, diagnostics, and industry, such as the classification of species through 16S rRNA and whole-genome sequencing. It is also fundamental in molecular cloning and genetic engineering, where bacterial DNA is manipulated to create recombinant DNA to express proteins in host cells. DNA extraction also allows the identification of antibiotic resistance genes, helping to track and combat the spread of resistant strains. As a diagnostic tool, bacterial DNA is essential for detecting pathogens through assays such as PCR (polymerase chain reaction), which are used in clinical, food safety, and environmental monitoring. The extraction of DNA is also a critical first step in metagenomics, enabling the study of microbial communities and their functions in various ecosystems. Additionally, bacterial DNA is used to study gene function and regulation, providing insights into bacterial physiology, metabolism, and adaptation. In biotechnology, purified DNA allows engineering bacteria for applications such as biosensors, biofuels, and the production of biomolecules. It also facilitates evolutionary and comparative genomics studies, shedding light on genetic variation, evolution, and horizontal gene transfer among bacteria. Other applications include vaccine development by identifying virulence factors and antigens, which can be targeted for preventing bacterial infections. In forensic microbiology, DNA helps trace contamination sources and analyze microbial evidence.
What are the steps, reagents and materials to extract DNA?
DNA extraction from cells involves isolating and purifying DNA from various types of tissues or cells. The process generally includes two main steps: (1) disruption of the cell membrane (and the cell wall, if present, as in plant cells) to release the DNA, and (2) purification, which involves separating the DNA from other cellular components and debris. First, the cell membrane is disrupted using a combination of the methods described in table 1 to get a fluid containing all the cell components, including the DNA. This process is called cell lysis and the resulted fluid is known as lysate (Figure 1).
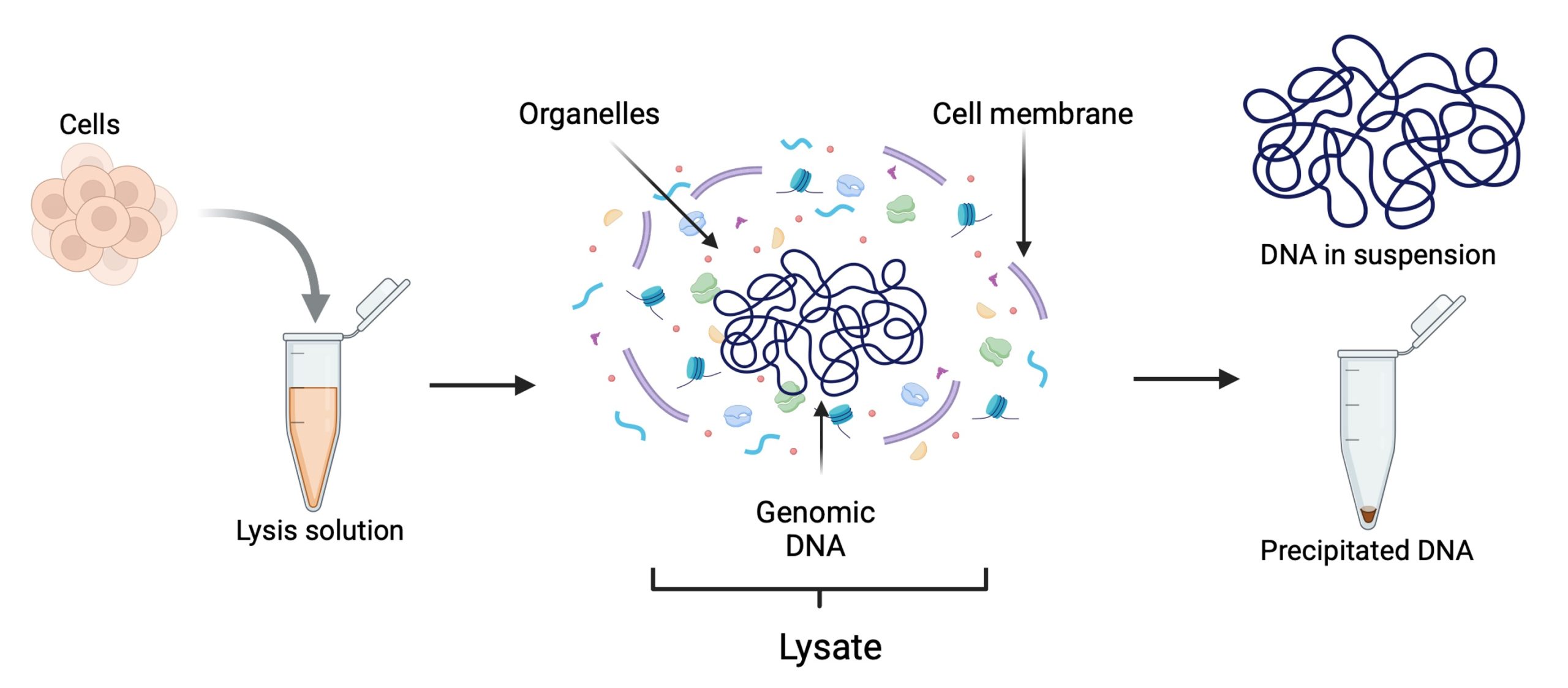
Figure 1. Cell lysis and lysate.
| Method | Description | Advantages | Disadvantages |
| Freeze-Thaw | Uses repeated freezing and thawing cycles to disrupt cell membranes through ice crystal formation and osmotic shock. | Simple, cost-effective, no special reagents required | Time-consuming; may cause enzyme denaturation. |
| Chemical | Employs chemicals (e.g., detergents, chaotropic agents) to solubilize cell membranes and denature proteins, allowing release of intracellular contents. | Effective for a wide range of cells; can be tailored to specific needs. | May require neutralization; can interfere with downstream assays. |
| Enzymatic | Uses enzymes (e.g., lysozyme, protease) to break down cell walls and membranes, often used for gentle lysis and preserving functional proteins. | Gentle on proteins and nucleic acids, preserving activity. | Specific to certain cell types; costly enzymes may be required. |
| Bead-Beating | Mechanical method using beads that physically grind and shear cells open, effective for tough cell walls such as bacteria, fungi, and plant cells. | Highly effective for hard-to-lyse cells; can process multiple samples simultaneously. | Requires specialized equipment; can cause sample heating and protein denaturation. |
| Sonication | Utilizes high-frequency sound waves to create cavitation bubbles that shear cells apart, effective for a wide range of cell types including bacteria and yeast. | Efficient and fast; scalable for small to large volumes. | May cause sample heating; requires specialized equipment and optimization. |
Table 1. Methods for cell disruption
During cell lysis, different reagents are used to break down different cellular components. Detergents and surfactants disrupt lipids, proteinases degrade proteins, and RNA is either enzymatically degraded or selectively removed through binding agents. Next, a concentrated salt solution is added to the lysate to aggregate the disrupted cell components, leaving the DNA dissolved in the solution. The lysate is then centrifuged at high speed or filtered through membranes to separate the DNA from the cellular debris. Finally, the DNA is precipitated, concentrated or bound to resins (Figure 2).
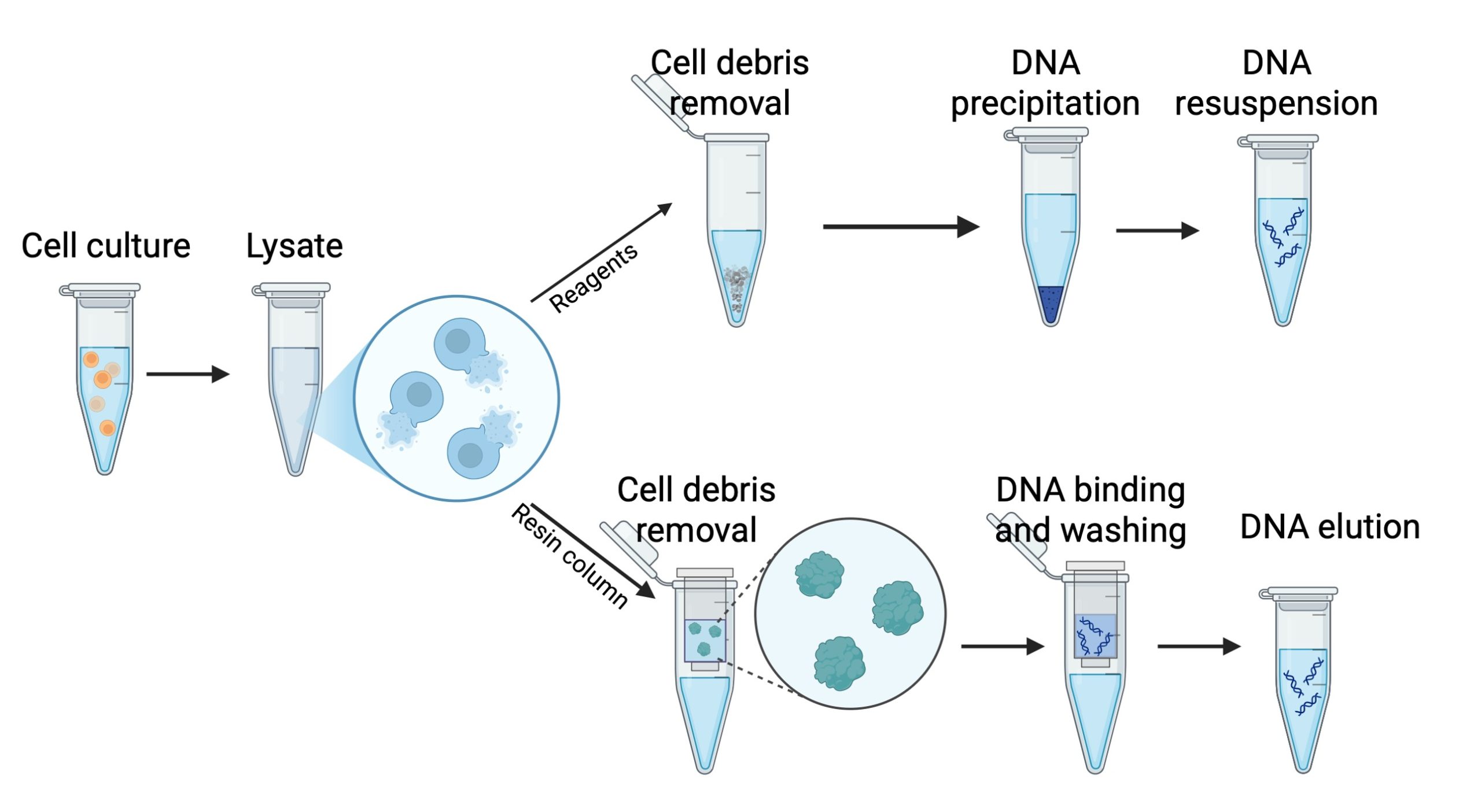
Figure 2. Reagent-based and column-based DNA purification approaches.
Composition and function of the different reagents commonly used for DNA extraction
- Tris/EDTA (TE Buffer)
- Acts as a buffering agent to maintain a stable pH (~ 8.0), which protects DNA from degradation during extraction.
- EDTA (Ethylenediamine-tetraacetic acid)
- Functions as a chelating agent that binds divalent metal ions like Mg²⁺ and Ca²⁺, essential cofactors for DNases (DNA-degrading enzymes).
- NaCl/KCl (Sodium Chloride/Potassium Chloride)
- Stabilize DNA by neutralizing the negative charges on the DNA phosphate backbone, reducing repulsion between DNA strands.
- Promote DNA precipitation during extraction and assist in removing proteins and contaminants by altering their solubility.
- Detergents (e.g., SDS, Triton X-100)
- Disrupt cell membranes by solubilizing lipids and proteins, lysing cells and releasing contents, including DNA.
- Denature proteins to ease their removal during purification.
- Reducing Agents (e.g., DTT, β-Mercaptoethanol)
- Break disulfide bonds in proteins, aiding in their denaturation and inactivating enzymes like DNase, which could degrade DNA.
- Enzymes (e.g., Proteinase K, RNase, Lysozyme)
- Proteinase K digests proteins, including those bound to DNA, aiding in their removal.
- RNase degrades RNA, ensuring RNA-free extracted material.
- Lysozyme breaks down bacterial cell walls, especially in gram-positive bacteria, to access DNA.
What are the steps to purify DNA from the lysate?
Ethanol Precipitation
Ethanol precipitation is widely used to purify and concentrate DNA from aqueous solutions. In this process, DNA is precipitated by adding ethanol (or isopropanol) and a salt, such as sodium acetate, which neutralizes the charges on the DNA phosphate backbone making it less soluble in water. The addition of cold ethanol causes the DNA to precipitate out of the solution, allowing it to be collected by centrifugation. This method is effective at removing small impurities, salts, and solvents from the DNA. It is cost-effective and suitable for precipitating DNA from large volumes, however it may co-precipitate some contaminants, such as proteins or polysaccharides, and requires careful handling to avoid DNA loss.
Phenol/Chloroform Extraction
Phenol/chloroform extraction is used to separate DNA from proteins and other cellular debris based on their solubility in organic solvents. The lysate is mixed with phenol and chloroform, creating a biphasic mixture. DNA remains in the aqueous phase, while proteins and lipids partition into the organic phase or interphase. After centrifugation, the aqueous phase containing is carefully collected and the DNA precipitated with ethanol, leaving behind impurities. This method is highly effective at removing proteins and other contaminants. It produces high-purity DNA with minimal protein contamination, but it uses toxic and hazardous chemicals that require careful handling and proper disposal, and it can be time-consuming.
Column Purification
Column purification uses silica-based columns that selectively bind DNA in the presence of high salt concentrations. The lysate is passed through the column, and DNA binds to the silica membrane while contaminants such as proteins, salts, and other impurities are washed away with specific buffers. Finally, the DNA is eluted from the column using a low-salt buffer or water. This method is highly efficient and produces high-quality DNA suitable for most molecular biology applications. This method is fast, easy to use, highly effective at removing contaminants, and compatible with automation for high-throughput processing. Most modern DNA extraction kits are column-based (Figure 3).
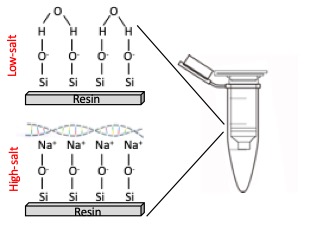
Figure 3. Column-based purification of DNA.
After purification, DNA is resuspended in TE buffer or pure water for use or storage.
What are the methods to measure the quantity and the quality of the extracted DNA?
Quantifying extracted DNA is essential to determine its concentration and purity for downstream applications like PCR, sequencing, and cloning. Some of the most commonly used methods for DNA quantification are:
UV Spectrophotometry (e.g. NanoDrop, nanophotometer)
UV spectrophotometry measures DNA concentration by estimating the absorbance at 260 nm in wavelength. The purity of DNA can also be assessed by measuring absorbance at 280 and 230 to estimate the concentration of proteins and salts/organic compounds, respectively, co-extracted with the DNA. This method is fast and simple, and it requires only a small amount of sample (as little as 1 µL). However, it cannot differentiate between DNA, RNA, and free nucleotides or contaminants that absorb at similar wavelengths.
Fluorometry (e.g. Qubit, PicoGreen, SYBR Green)
Fluorometric methods quantify DNA using fluorescent dyes that specifically bind to DNA (or RNA), such as PicoGreen or Qubit reagents. The fluorescence intensity, measured by a fluorometer, is proportional to the DNA concentration in the sample. This method is highly specific and sensitive and it is less affected by contaminants compared to spectrophotometry. It requires specific reagents and equipment and does not provide information on the purity or size of the DNA.
Capillary Electrophoresis (e.g., Bioanalyzer, TapeStation)
Capillary electrophoresis separates DNA fragments by size and measures their concentration using fluorescent dyes. The system provides quantitative data on DNA concentration and fragment size.
Agarose Gel Electrophoresis stained with ethidium bromide or SYBR green.
The DNA is run on an agarose gel and stained with dyes like ethidium bromide or SYBR green, which fluoresce under UV or blue light. The intensity of the fluorescence of the DNA bands can be compared to a DNA ladder of known concentrations to estimate the DNA quantity. This method allows the visualization of DNA quality, size distribution, and integrity in addition to quantification. It is not very accurate for precise quantification; requires gel electrophoresis setup, staining, and imaging equipment. It is also less sensitive compared to spectrophotometry and fluorometry. The preparation of an agarose gel for DNA electrophoresis requires the use of buffers such as TAE (Tris-acetic acid-EDTA) or TBE (Tris-boric acid-EDTA).
- TBE (Tris-borate-EDTA) is a better conductive medium than TAE (Tris-acetate EDTA) and so it is less prone to overheating during long runs (> 2 h).
- Borate is an enzyme inhibitor so TBE is not a good buffer to use if you will be isolating the DNA for downstream enzymatic steps. For example, borate carry-over could affect your ligations, so use TAE instead.
- Acetate gives improved separation of large DNA fragments
- On the other hand, borate resolves <2kb fragments better, so use TBE for smaller fragments.
 Figure 4. A) Genomic DNA on a 1% agarose gel stained with SYBR green, B) Wavelengths used to estimate DNA concentration and purity.
Figure 4. A) Genomic DNA on a 1% agarose gel stained with SYBR green, B) Wavelengths used to estimate DNA concentration and purity.
Molecular weight markers
Also known as DNA ladders, MWM are a mixture of DNA molecules of different lengths used in agarose or acrylamide gel electrophoresis to be used as a reference to estimate the size of unknown DNA molecules separated on the basis of their mobility in an electrical field through the gel. DNA ladders are routinely used in every molecular biology laboratory and they are formulated for different applications. For instance, 100 bp ladders are used along the products of PCR reactions, while 1 kb ladders are preferred for genomic DNA extractions.

Figure 5. DNA weight markers or DNA “ladders”
DNA extraction from an overnight E. coli culture
Materials
Micropipettors set
Sterile micropipettor tips
1.7 mL Eppendorf tubes
Tubes rack
Vortex
Microcentrifuge
Cell disruptor (or tube shaking adapter for vortex)
Quick-DNA fungal/bacterial miniprep kit from Zymo Research https://www.zymoresearch.com/collections/quick-dna-fungal-bacterial-kits/products/quick-dna-fungal-bacterial-miniprep-kit
Safety concerns
This DNA extraction uses chemicals, biological materials, and equipment that pose some potential risks:
- Handle the ethanol and the kit reagents with gloves, at all times.
- Use biosafety protocols to handle the bacteria samples to prevent accidental exposure, and dispose of all the waste in a biohazard container.
- Make sure you balance the microcentrifuges correctly before you use them.
- Avoid eating or drinking, and manage spills and emergencies following the directions of the instructor.
Protocol
DNA extraction from Escherichia coli cultures
- Transfer 1.7 mL of an E. coli overnight culture into an Eppendorf tube and centrifuge at 10000 rpm for 5 min. Discard the supernatant.
- Resuspend the cell pellet in 200 uL of 1X PBS and transfer to a ZR BashingBead Lysis tube.
- Add 750 uL of BashingBead buffer and cap tightly.
- Secure the tubes in a bead beater and process at maximum speed for 20 min.
- Centrifuge the lysis tube at 10000 rpm for 1 min.
- Transfer 400 uL of the supernatant to a Zymo-Spin III-F filter in a collection tube and centrifuge at 8000 rpm for 1 min. Discard the filter.
- Add 1200 uL of genomic lysis buffer to the filtrate in the collection tube and mix well with a pipette.
- Transfer 800 uL of the mixture from the previous step into a Zymo-Spin IICR column in a collection tube and centrifuge at 10000 rpm for 1 min.
- Discard the flow through from the collection tube ad repeat the previous step. Discard the flow through.
- Add 200 uL of DNA pre-wash buffer to the IICR column in a new collection tube and centrifuge at 10000 rpm for 1 min. Discard the flow through.
- Add 500 uL of g-DNA wash buffer to the IICR column and centrifuge at 10000 rpm for 1 min.
- Transfer the IICR column to a clean 1.5 mL microcentrifuge tube and add 50 uL of DNA elution buffer directly into the column matrix an incubate at room temperature 1 min. Centrifuge at 10000 rpm for 30 sec to elute the DNA.
- Label the tube and keep frozen until use.
Preparing, pouring and running an agarose gel
- To prepare a 1% agarose gel, weigh either 0.7 or 1 g of agarose (for a small and large gel, respectively) and transfer it into a 200 mL beaker with either 70 or 100 mL of 1X TBE buffer. Heat the mixture in the microwave in 30 s increments, hand stirring the content gently with a circular motion until the agarose dissolves completely. Once The agarose is completely dissolved, add either 7 or 10 uL of SYBR green, dissolve it and pour the mix into the gel bed and insert the comb on the negative side of the chamber (Figure 6A). Let it solidify (~20 min) or until it turns opaque.
- Once the gel solidifies, place it in the electrophoresis chamber and pour 1X TBE buffer on both sides of the tank until the gel is covered with a ~2 mm thick buffer layer. Pull out the comb gently without breaking the walls of the wells.
- Take the DNA samples out of the freezer and thaw them at room temperature. Mix 10 uL of the DNA with 5 uL of loading buffer on a 2 X 2 cm piece of parafilm paper, pipetting the liquids up and down twice. Skipping the first and last lanes of the gel, pipet the mix (~15 uL) into separate wells of the gel (Figure 6B). Save the first and last lanes of each gel for the DNA molecular weight marker or DNA ladder)
- Connect the electrodes of the chamber to the power supply (make sure the DNA samples are on the negative side!) and run at 100-120 mV for 40-60 m or until the blue dye in the loading buffer reaches ¾ of the length of the gel (Figure 6C). Once the run is complete, place the gel in the transilluminator and follow the instructor directions to take a photograph for your lab report.
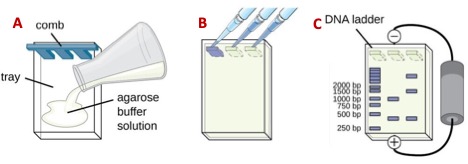
Figure 6. A) pouring, B) loading, and C) running a DNA agarose gel.
Key Takeaways
-
DNA extraction is used to isolate DNA from cells (bacteria, mammalian and plant cells, tissues, soil, food, etc) so it can be analyzed for genetic studies, diagnostics, or for biotechnology applications.
-
The extraction process typically includes cell lysis, removal of proteins and other contaminants, and purification of the DNA—using lysis buffer, enzymes or detergents, and alcohol or resins, respectively.
-
The quality and yield of DNA depend heavily on minimizing contamination and on pipetting accurate volumes of the different reagents. The information obtained from the spectrophotometric analysis at 260 280 nm in wavelength and from electrophoresis is essential to determine its suitability for further experiments.
Lecture slides
Download the slides to follow the pre-lab lecture. DNAextraction_2025
Lab report
Download the DNA extraction lab report file and submit on Canvas, following the directions of your instructor.
PCR (Polymerase Chain Reaction) is a technique used to amplify specific DNA sequences, generating millions of copies from a small DNA sample through repeated cycles of heating and cooling.
Metagenomics is the study of genetic material recovered directly from environmental samples, allowing scientists to analyze entire communities of microorganisms—such as bacteria, viruses, fungi, and other microbes—without the need to culture them in a lab. This approach is particularly powerful for understanding complex ecosystems, as it captures the genetic diversity of microbes in their natural habitats, such as soil, oceans, or the human gut.
A lysate is a solution containing the contents of lysed (broken open) cells. When cells are lysed, their membranes are disrupted, releasing internal components such as proteins, nucleic acids, and organelles into the surrounding solution.
To prepare 1 liter of 1X TAE buffer, you’ll need the following:
Tris Base: 4.84 grams (40 mM)
Acetic Acid: 5.71 milliliters of glacial acetic acid (40 mM)
EDTA (0.5 M, pH 8.0 stock): 2 milliliters (1 mM final concentration)
Here's the step-by-step recipe:
Add 4.84 g of Tris base to a container.
Add 5.71 ml of glacial acetic acid.
Add 2 ml of 0.5 M EDTA (pH 8.0) solution.
Fill with distilled water up to 1 liter.
Mix until fully dissolved and adjust the pH to 8.0 if necessary.
Tris Base: 10.8 grams (89 mM)
Boric Acid: 5.5 grams (89 mM)
EDTA (0.5 M, pH 8.0 stock): 2 milliliters (1 mM final concentration)
Here’s the step-by-step recipe:
Add 10.8 g of Tris base to a container.
Add 5.5 g of boric acid.
Add 2 ml of 0.5 M EDTA (pH 8.0) solution.
Fill with distilled water up to 1 liter.
Mix until all components are fully dissolved.
Potassium Chloride (KCl): 0.2 grams
Disodium Phosphate (Na₂HPO₄): 1.44 grams
Monopotassium Phosphate (KH₂PO₄): 0.24 grams
Here’s the step-by-step recipe:
Add 8 g of NaCl to a container.
Add 0.2 g of KCl.
Add 1.44 g of Na₂HPO₄.
Add 0.24 g of KH₂PO₄.
Fill with distilled water up to 1 liter.
Adjust the pH to 7.4 if needed.
3000 rpm if using the VortexGenie with the upright tubes adaptor.
The clear liquid that separates from the solids AFTER you centrifuge the sample.